









 Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them.
Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them. The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle.
The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle. The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
Gemin 1 Polyclonal Antibody
Purified Rabbit Polyclonal Antibody (Pab)
- SPECIFICATION
- CITATIONS
- PROTOCOLS
- BACKGROUND

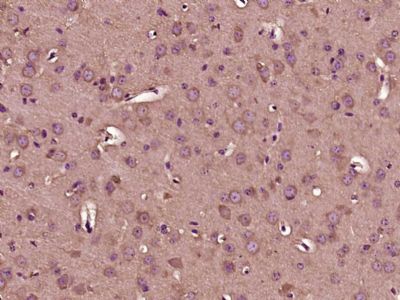
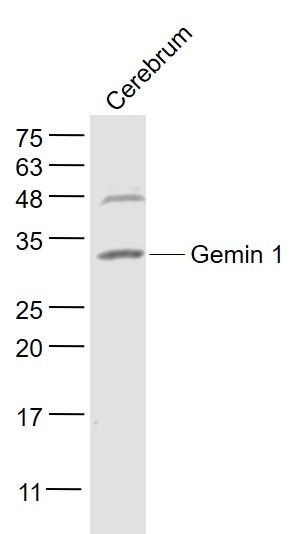
Application
| WB, IHC-P, IHC-F, IF, ICC, E |
|---|---|
| Primary Accession | Q16637 |
| Reactivity | Rat, Pig, Dog, Bovine |
| Host | Rabbit |
| Clonality | Polyclonal |
| Calculated MW | 31 KDa |
| Physical State | Liquid |
| Immunogen | KLH conjugated synthetic peptide derived from human Gemin 1 |
| Epitope Specificity | 31-100/294 |
| Isotype | IgG |
| Purity | affinity purified by Protein A |
| Buffer | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| SUBCELLULAR LOCATION | Cytoplasm. Nucleus, gem. Note=Localized in subnuclear structures next to coiled bodies, called Gemini of Cajal bodies (Gems). |
| SIMILARITY | Belongs to the SMN family. Contains 1 Tudor domain. |
| SUBUNIT | Component of an import snRNP complex composed of KPNB1, RNUT1, SMN1 and ZNF259. Part of the core SMN complex that contains SMN1, GEMIN2/SIP1, DDX20/GEMIN3, GEMIN4, GEMIN5, GEMIN6, GEMIN7, GEMIN8 and STRAP/UNRIP. Interacts with DDX20, FBL, NOLA1, RNUT1, SYNCRIP and with several spliceosomal snRNP core Sm proteins, including SNRPB, SNRPD1, SNRPD2, SNRPD3, SNRPE and ILF3. Interacts with OSTF1, LSM10 and LSM11. |
| DISEASE | Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 1 (SMA1) [MIM:253300]. Spinal muscular atrophy refers to a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. Autosomal recessive forms are classified according to the age of onset, the maximum muscular activity achieved, and survivorship. The severity of the disease is mainly determined by the copy number of SMN2, a copy gene which predominantly produces exon 7-skipped transcripts and only low amount of full-length transcripts that encode for a protein identical to SMN1. Only about 4% of SMA patients bear one SMN1 copy with an intragenic mutation. SMA1 is a severe form, with onset before 6 months of age. SMA1 patients never achieve the ability to sit. Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 2 (SMA2) [MIM:253550]. SMA2 is an autosomal recessive spinal muscular atrophy of intermediate severity, with onset between 6 and 18 months. Patients do not reach the motor milestone of standing, and survive into adulthood. Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 3 (SMA3) [MIM:253400]. SMA3 is an autosomal recessive spinal muscular atrophy with onset after 18 months. SMA3 patients develop ability to stand and walk and survive into adulthood. Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 4 (SMA4) [MIM:271150]. SMA4 is an autosomal recessive spinal muscular atrophy characterized by symmetric proximal muscle weakness with onset in adulthood and slow disease progression. SMA4 patients can stand and walk. |
| Important Note | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| Background Descriptions | This gene is part of a 500 kb inverted duplication on chromosome 5q13. This duplicated region contains at least four genes and repetitive elements which make it prone to rearrangements and deletions. The repetitiveness and complexity of the sequence have also caused difficulty in determining the organization of this genomic region. The telomeric and centromeric copies of this gene are nearly identical and encode the same protein. However, mutations in this gene, the telomeric copy, are associated with spinal muscular atrophy; mutations in the centromeric copy do not lead to disease. The centromeric copy may be a modifier of disease caused by mutation in the telomeric copy. The critical sequence difference between the two genes is a single nucleotide in exon 7, which is thought to be an exon splice enhancer. Note that the nine exons of both the telomeric and centromeric copies are designated historically as exon 1, 2a, 2b, and 3-8. It is thought that gene conversion events may involve the two genes, leading to varying copy numbers of each gene. The protein encoded by this gene localizes to both the cytoplasm and the nucleus. Within the nucleus, the protein localizes to subnuclear bodies called gems which are found near coiled bodies containing high concentrations of small ribonucleoproteins (snRNPs). This protein forms heteromeric complexes with proteins such as SIP1 and GEMIN4, and also interacts with several proteins known to be involved in the biogenesis of snRNPs, such as hnRNP U protein and the small nucleolar RNA binding protein. Two transcript variants encoding distinct isoforms have been described. [provided by RefSeq, Sep 2008] |
| Gene ID | 6606;6607 |
|---|---|
| Other Names | Survival motor neuron protein, Component of gems 1, Gemin-1, SMN1, SMN, SMNT |
| Target/Specificity | Expressed in a wide variety of tissues. Expressed at high levels in brain, kidney and liver, moderate levels in skeletal and cardiac muscle, and low levels in fibroblasts and lymphocytes. Also seen at high levels in spinal cord. Present in osteoclasts and mononuclear cells (at protein level). |
| Dilution | WB=1:500-2000,IHC-P=1:100-500,IHC-F=1:100-500,ICC=1:100-500,IF=1:100-500,ELISA=1:5000-10000 |
| Storage | Store at -20 ℃ for one year. Avoid repeated freeze/thaw cycles. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 ℃. |
| Name | SMN1 |
|---|---|
| Synonyms | SMN, SMNT |
| Function | The SMN complex catalyzes the assembly of small nuclear ribonucleoproteins (snRNPs), the building blocks of the spliceosome, and thereby plays an important role in the splicing of cellular pre- mRNAs (PubMed:18984161, PubMed:9845364). Most spliceosomal snRNPs contain a common set of Sm proteins SNRPB, SNRPD1, SNRPD2, SNRPD3, SNRPE, SNRPF and SNRPG that assemble in a heptameric protein ring on the Sm site of the small nuclear RNA to form the core snRNP (Sm core) (PubMed:18984161). In the cytosol, the Sm proteins SNRPD1, SNRPD2, SNRPE, SNRPF and SNRPG are trapped in an inactive 6S pICln-Sm complex by the chaperone CLNS1A that controls the assembly of the core snRNP (PubMed:18984161). To assemble core snRNPs, the SMN complex accepts the trapped 5Sm proteins from CLNS1A forming an intermediate (PubMed:18984161). Within the SMN complex, SMN1 acts as a structural backbone and together with GEMIN2 it gathers the Sm complex subunits (PubMed:17178713, PubMed:21816274, PubMed:22101937). Binding of snRNA inside 5Sm ultimately triggers eviction of the SMN complex, thereby allowing binding of SNRPD3 and SNRPB to complete assembly of the core snRNP (PubMed:31799625). Ensures the correct splicing of U12 intron- containing genes that may be important for normal motor and proprioceptive neurons development (PubMed:23063131). Also required for resolving RNA-DNA hybrids created by RNA polymerase II, that form R- loop in transcription terminal regions, an important step in proper transcription termination (PubMed:26700805). May also play a role in the metabolism of small nucleolar ribonucleoprotein (snoRNPs). |
| Cellular Location | Nucleus, gem. Nucleus, Cajal body. Cytoplasm. Cytoplasmic granule. Perikaryon. Cell projection, neuron projection. Cell projection, axon {ECO:0000250|UniProtKB:P97801}. Cytoplasm, myofibril, sarcomere, Z line {ECO:0000250|UniProtKB:P97801}. Note=Colocalizes with actin and at the Z-line of skeletal muscle (By similarity). Under stress conditions colocalizes with RPP20/POP7 in punctuated cytoplasmic granules (PubMed:14715275). Colocalized and redistributed with ZPR1 from the cytoplasm to nuclear gems (Gemini of coiled bodies) and Cajal bodies (PubMed:11283611). Colocalizes with FMR1 in cytoplasmic granules in the soma and neurite cell processes (PubMed:18093976) {ECO:0000250|UniProtKB:P97801, ECO:0000269|PubMed:11283611, ECO:0000269|PubMed:14715275, ECO:0000269|PubMed:18093976} |
| Tissue Location | Expressed in a wide variety of tissues. Expressed at high levels in brain, kidney and liver, moderate levels in skeletal and cardiac muscle, and low levels in fibroblasts and lymphocytes. Also seen at high levels in spinal cord. Present in osteoclasts and mononuclear cells (at protein level). |
Research Areas
Citations (0)

Thousands of laboratories across the world have published research that depended on the performance of antibodies from Abcepta to advance their research. Check out links to articles that cite our products in major peer-reviewed journals, organized by research category.
Submit your citation using an Abcepta antibody to
info@abcepta.com, and receive a free "I Love Antibodies" mug.
info@abcepta.com, and receive a free "I Love Antibodies" mug.
Application Protocols
Provided below are standard protocols that you may find useful for product applications.
Abcepta welcomes feedback from its customers.
If you have used an Abcepta product and would like to share how it has performed, please click on the "Submit Review" button and provide the requested information. Our staff will examine and post your review and contact you if needed.
If you have any additional inquiries please email technical services at tech@abcepta.com.
$ 385.00
Cat# AP54544
Ordering Information
United States
AlbaniaAustraliaAustriaBelgiumBosnia & HerzegovinaBrazilBulgariaCanadaCentral AmericaChinaCroatiaCyprusCzech RepublicDenmarkEstoniaFinlandFranceGermanyGreeceHong KongHungaryIcelandIndiaIndonesiaIrelandIsraelItalyJapanLatviaLithuaniaLuxembourgMacedoniaMalaysiaMaltaMexicoNetherlandsNew ZealandNorwayPakistanPolandPortugalRomaniaSerbiaSingaporeSlovakiaSloveniaSouth AfricaSouth KoreaSpainSwedenSwitzerlandTaiwanTurkeyUnited KingdomUnited StatesVietnamWorldwideOthers
USA Headquarters
(888) 735-7227 / (858) 622-0099 or (858) 875-1900
Other Products
Shipping Information
Domestic orders (in stock items)
Shipped out the same day. Orders placed after 1 PM (PST) will ship out the next business day.
International orders
Contact your local distributors
