


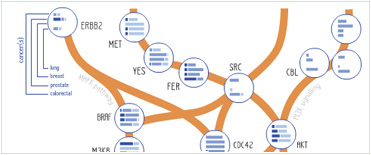







 Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them.
Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them. The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle.
The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle. The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
GDAP1 Polyclonal Antibody
Purified Rabbit Polyclonal Antibody (Pab)

- SPECIFICATION
- CITATIONS
- PROTOCOLS
- BACKGROUND

Application
| WB, IHC-P, IHC-F, IF, ICC, E |
|---|---|
| Primary Accession | Q8TB36 |
| Reactivity | Rat, Pig, Dog, Bovine |
| Host | Rabbit |
| Clonality | Polyclonal |
| Calculated MW | 41 KDa |
| Physical State | Liquid |
| Immunogen | KLH conjugated synthetic peptide derived from human GDAP1 |
| Epitope Specificity | 151-230/358 |
| Isotype | IgG |
| Purity | affinity purified by Protein A |
| Buffer | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| SUBCELLULAR LOCATION | Mitochondrion outer membrane; Multi-pass membrane protein. Cytoplasm (By similarity). |
| SIMILARITY | Belongs to the GST superfamily. Contains 1 GST C-terminal domain. Contains 1 GST N-terminal domain. |
| SUBUNIT | Homodimer. |
| DISEASE | Defects in GDAP1 are the cause of Charcot-Marie-Tooth disease type 4A (CMT4A) [MIM:214400]. CMT4A is a form of Charcot-Marie-Tooth disease, the most common inherited disorder of the peripheral nervous system. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathy or CMT1, and primary peripheral axonal neuropathy or CMT2. Demyelinating CMT neuropathies are characterized by severely reduced nerve conduction velocities (less than 38 m/sec), segmental demyelination and remyelination with onion bulb formations on nerve biopsy, slowly progressive distal muscle atrophy and weakness, absent deep tendon reflexes, and hollow feet. Autosomal recessive forms of demyelinating Charcot-Marie-Tooth disease are by convention designated CMT4. CMT4A is a severe form characterized by early age of onset and rapid progression leading to inability to walk in late childhood or adolescence. Defects in GDAP1 are the cause of Charcot-Marie-Tooth disease axonal recessive with vocal cord paresis (CMT2RV) [MIM:607706]. CMT2RV is a form of Charcot-Marie-Tooth disease characterized by the association of axonal neuropathy with vocal cord paresis. Defects in GDAP1 are the cause of Charcot-Marie-Tooth disease type 2K (CMT2K) [MIM:607831]. CMT2K is an axonal form of Charcot-Marie-Tooth disease. Axonal CMT neuropathies are characterized by signs of axonal regeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2K onset is in early childhood (younger than 3 years). This phenotype is characterized by foot deformities, kyphoscoliosis, distal limb muscle weakness and atrophy, areflexia, and diminished sensation in the lower limbs. Weakness in the upper limbs is observed in the first decade, with clawing of the fingers. Inheritance can be autosomal dominant or recessive. Defects in GDAP1 are the cause of Charcot-Marie-Tooth disease recessive intermediate type A (CMTRIA) [MIM:608340]. CMTRIA is a form of Charcot-Marie-Tooth disease characterized by clinical and pathologic features intermediate between demyelinating and axonal peripheral neuropathies, and motor median nerve conduction velocities ranging from 25 to 45 m/sec. |
| Important Note | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| Background Descriptions | Glutathione S-transferases (GSTs) function to conjugate reduced glutathione to many exogenous and endogenous hydrophobic electrophiles. Although it shares the carboxy and amino-terminal glutathione S-transferase domains, GDAP1 is characterized as a GST-like protein because it contains an extended GST domain II and a predicted transmembrane domain, two characteristics which are unusual for GST family members. GDAP1 may function in a signal transduction pathway that is responsible for ganglioside-induced neurite differentiation and also may play a role in protecting myelin membranes from free-radical damage. Mutations in the gene encoding GDAP1 is the cause of many forms of Charcot-Marie-Tooth disease, a common inherited disorder of the peripheral nervous system that is characterized by reduced nerve conduction velocities, slow progressive distal muscle atrophy and absent deep tendon reflexes. |
| Gene ID | 54332 |
|---|---|
| Other Names | Ganglioside-induced differentiation-associated protein 1, GDAP1, GDAP1 |
| Target/Specificity | Highly expressed in whole brain and spinal cord. Predominant expression in central tissues of the nervous system not only in neurons but also in Schwann cells. |
| Dilution | WB=1:500-2000,IHC-P=1:100-500,IHC-F=1:100-500,ICC=1:100-500,IF=1:100-500,ELISA=1:5000-10000 |
| Storage | Store at -20 ℃ for one year. Avoid repeated freeze/thaw cycles. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 ℃. |
| Name | GDAP1 |
|---|---|
| Function | Regulates the mitochondrial network by promoting mitochondrial fission. |
| Cellular Location | Mitochondrion outer membrane; Multi-pass membrane protein. Cytoplasm {ECO:0000250|UniProtKB:O88741} |
| Tissue Location | Highly expressed in whole brain and spinal cord. Predominant expression in central tissues of the nervous system not only in neurons but also in Schwann cells |
Research Areas
Citations (0)

Thousands of laboratories across the world have published research that depended on the performance of antibodies from Abcepta to advance their research. Check out links to articles that cite our products in major peer-reviewed journals, organized by research category.
Submit your citation using an Abcepta antibody to
info@abcepta.com, and receive a free "I Love Antibodies" mug.
info@abcepta.com, and receive a free "I Love Antibodies" mug.
Application Protocols
Provided below are standard protocols that you may find useful for product applications.
Abcepta welcomes feedback from its customers.
If you have used an Abcepta product and would like to share how it has performed, please click on the "Submit Review" button and provide the requested information. Our staff will examine and post your review and contact you if needed.
If you have any additional inquiries please email technical services at tech@abcepta.com.
$ 385.00
Cat# AP54610
Ordering Information
United States
AlbaniaAustraliaAustriaBelgiumBosnia & HerzegovinaBrazilBulgariaCanadaCentral AmericaChinaCroatiaCyprusCzech RepublicDenmarkEstoniaFinlandFranceGermanyGreeceHong KongHungaryIcelandIndiaIndonesiaIrelandIsraelItalyJapanLatviaLithuaniaLuxembourgMacedoniaMalaysiaMaltaMexicoNetherlandsNew ZealandNorwayPakistanPolandPortugalRomaniaSerbiaSingaporeSlovakiaSloveniaSouth AfricaSouth KoreaSpainSwedenSwitzerlandTaiwanTurkeyUnited KingdomUnited StatesVietnamWorldwideOthers
USA Headquarters
(888) 735-7227 / (858) 622-0099 or (858) 875-1900
Other Products
Shipping Information
Domestic orders (in stock items)
Shipped out the same day. Orders placed after 1 PM (PST) will ship out the next business day.
International orders
Contact your local distributors
