


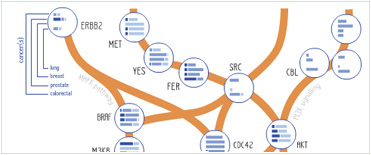







 Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them.
Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them. The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle.
The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle. The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
FKRP Polyclonal Antibody
Purified Rabbit Polyclonal Antibody (Pab)
- SPECIFICATION
- CITATIONS
- PROTOCOLS
- BACKGROUND

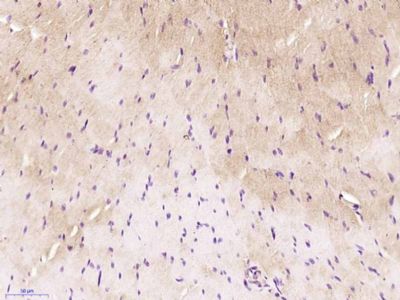
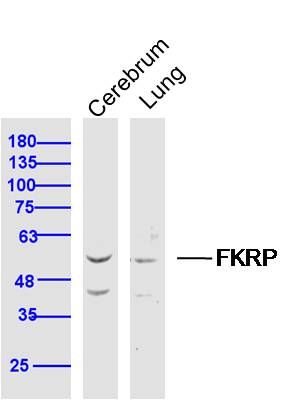
Application
| WB, IHC-P, IHC-F, IF, ICC, E |
|---|---|
| Primary Accession | Q9H9S5 |
| Reactivity | Rat, Pig, Dog |
| Host | Rabbit |
| Clonality | Polyclonal |
| Calculated MW | 55 KDa |
| Physical State | Liquid |
| Immunogen | KLH conjugated synthetic peptide derived from human FKRP |
| Epitope Specificity | 1-100/495 |
| Purity | affinity purified by Protein A |
| Buffer | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| SUBCELLULAR LOCATION | Golgi apparatus. Secreted. Cell membrane > sarcolemma. Rough endoplasmic reticulum. According to some studies the N-terminal hydrophobic domain is cleaved after translocation to the Golgi apparatus and the protein is secreted. According to others the N-terminal hydrophobic domain is a transmembrane domain and the protein is a type II transmembrane type targeted to the Golgi apparatus by a non-cleavable signal anchor sequence. Localization at the cell membrane may require the presence of dystroglycan. At the Golgi apparatus localizes most likely at the cis-compartment. Detected in rough endoplasmic reticulum in myocytes. In general, mutants associated with severe clinical phenotypes are retained within the endoplasmic reticulum. |
| SIMILARITY | Belongs to the licD transferase family. |
| Post-translational modifications | N-glycosylated. |
| DISEASE | Defects in FKRP are the cause of muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies type A5 (MDDGA5) [MIM:613153]. MDDGA5 is an autosomal recessive disorder characterized by congenital muscular dystrophy associated with cobblestone lissencephaly and other brain anomalies, eye malformations, profound mental retardation, and death usually in the first years of life. Included diseases are the more severe Walker-Warburg syndrome and the slightly less severe muscle-eye-brain disease. Defects in FKRP are the cause of muscular dystrophy-dystroglycanopathy congenital with or without mental retardation type B5 (MDDGB5) [MIM:606612]. MDDGB5 is a congenital muscular dystrophy characterized by a severe phenotype with inability to walk, muscle hypertrophy, marked elevation of serum creatine kinase, a secondary deficiency of laminin alpha2, and a marked reduction in alpha-dystroglycan expression. Only a subset of MDDGB5 patients have brain involvements. Defects in FKRP are the cause of muscular dystrophy-dystroglycanopathy limb-girdle type C5 (MDDGC5) [MIM:607155]; also known as limb-girdle muscular dystrophy type 2I. MDDGC5 is an autosomal recessive disorder with age of onset ranging from childhood to adult life, and variable severity. Clinical features include proximal muscle weakness, waddling gait, calf hypertrophy, cardiomyopathy and respiratory insufficiency. A reduction of alpha-dystroglycan and laminin alpha-2 expression can be observed on skeletal muscle biopsy from MDDGC5 patients. |
| Important Note | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| Background Descriptions | This gene encodes a protein which is targeted to the medial Golgi apparatus and is necessary for posttranslational modification of dystroglycan. Mutations in this gene have been associated with congenital muscular dystrophy, mental retardation, and cerebellar cysts. Several alternatively spliced transcript variants of this gene have been described, but the full-length nature of some of these variants has not been determined. [provided by RefSeq, Oct 2008] |
| Gene ID | 79147 |
|---|---|
| Other Names | Fukutin-related protein, 2.4.2.-, Ribitol-5-phosphate transferase, FKRP |
| Target/Specificity | Expressed predominantly in skeletal muscle, placenta, and heart and relatively weakly in brain, lung, liver kidney and pancreas. |
| Dilution | WB=1:500-2000,IHC-P=1:100-500,IHC-F=1:100-500,ICC=1:100-500,IF=1:100-500,ELISA=1:5000-10000 |
| Storage | Store at -20 ℃ for one year. Avoid repeated freeze/thaw cycles. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 ℃. |
| Name | FKRP (HGNC:17997) |
|---|---|
| Function | Catalyzes the transfer of a ribitol 5-phosphate from CDP-L- ribitol to the ribitol 5-phosphate previously attached by FKTN/fukutin to the phosphorylated O-mannosyl trisaccharide (N-acetylgalactosamine- beta-3-N-acetylglucosamine-beta-4-(phosphate-6-)mannose), a carbohydrate structure present in alpha-dystroglycan (DAG1) (PubMed:26923585, PubMed:27194101, PubMed:29477842, PubMed:31949166). This constitutes the second step in the formation of the ribose 5- phosphate tandem repeat which links the phosphorylated O-mannosyl trisaccharide to the ligand binding moiety composed of repeats of 3- xylosyl-alpha-1,3-glucuronic acid-beta-1 (PubMed:25279699, PubMed:26923585, PubMed:27194101, PubMed:29477842, PubMed:31949166). |
| Cellular Location | Golgi apparatus membrane; Single-pass type II membrane protein. Secreted. Cell membrane, sarcolemma {ECO:0000250|UniProtKB:Q8CG64}. Rough endoplasmic reticulum. Cytoplasm {ECO:0000250|UniProtKB:Q8CG64}. Note=According to some studies the N- terminal hydrophobic domain is cleaved after translocation to the Golgi apparatus and the protein is secreted (PubMed:19900540). Localization at the cell membrane may require the presence of dystroglycan (By similarity). At the Golgi apparatus localizes to the middle-to-trans- cisternae, as assessed by MG160 colocalization. Detected in rough endoplasmic reticulum in myocytes (PubMed:17554798, PubMed:21886772) In general, mutants associated with severe clinical phenotypes are retained within the endoplasmic reticulum (PubMed:15213246) {ECO:0000250|UniProtKB:Q8CG64, ECO:0000269|PubMed:15213246, ECO:0000269|PubMed:17554798, ECO:0000269|PubMed:19900540, ECO:0000269|PubMed:21886772} |
| Tissue Location | Expressed in the retina (at protein level) (PubMed:29416295). Expressed predominantly in skeletal muscle, placenta, and heart and relatively weakly in brain, lung, liver, kidney, and pancreas (PubMed:11592034). |
Research Areas
Citations (0)

Thousands of laboratories across the world have published research that depended on the performance of antibodies from Abcepta to advance their research. Check out links to articles that cite our products in major peer-reviewed journals, organized by research category.
Submit your citation using an Abcepta antibody to
info@abcepta.com, and receive a free "I Love Antibodies" mug.
info@abcepta.com, and receive a free "I Love Antibodies" mug.
Application Protocols
Provided below are standard protocols that you may find useful for product applications.
Abcepta welcomes feedback from its customers.
If you have used an Abcepta product and would like to share how it has performed, please click on the "Submit Review" button and provide the requested information. Our staff will examine and post your review and contact you if needed.
If you have any additional inquiries please email technical services at tech@abcepta.com.
$ 385.00
Cat# AP56118
Ordering Information
United States
AlbaniaAustraliaAustriaBelgiumBosnia & HerzegovinaBrazilBulgariaCanadaCentral AmericaChinaCroatiaCyprusCzech RepublicDenmarkEstoniaFinlandFranceGermanyGreeceHong KongHungaryIcelandIndiaIndonesiaIrelandIsraelItalyJapanLatviaLithuaniaLuxembourgMacedoniaMalaysiaMaltaMexicoNetherlandsNew ZealandNorwayPakistanPolandPortugalRomaniaSerbiaSingaporeSlovakiaSloveniaSouth AfricaSouth KoreaSpainSwedenSwitzerlandTaiwanTurkeyUnited KingdomUnited StatesVietnamWorldwideOthers
USA Headquarters
(888) 735-7227 / (858) 622-0099 or (858) 875-1900
Other Products
Shipping Information
Domestic orders (in stock items)
Shipped out the same day. Orders placed after 1 PM (PST) will ship out the next business day.
International orders
Contact your local distributors
