


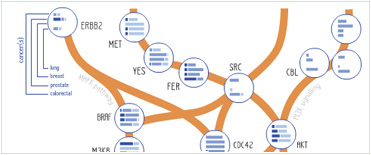







 Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them.
Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them. The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle.
The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle. The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
GCS1 Antibody (N-term) Blocking Peptide
Synthetic peptide
- SPECIFICATION
- CITATIONS
- PROTOCOLS
- BACKGROUND
| Primary Accession | Q13724 |
|---|---|
| Other Accession | NP_006293 |
| Clone Names | 3103127 |
| Gene ID | 7841 |
|---|---|
| Other Names | Mannosyl-oligosaccharide glucosidase, Processing A-glucosidase I, MOGS, GCS1 |
| Target/Specificity | The synthetic peptide sequence used to generate the antibody AP2315a was selected from the N-term region of human GCS1 . A 10 to 100 fold molar excess to antibody is recommended. Precise conditions should be optimized for a particular assay. |
| Format | Peptides are lyophilized in a solid powder format. Peptides can be reconstituted in solution using the appropriate buffer as needed. |
| Storage | Maintain refrigerated at 2-8°C for up to 6 months. For long term storage store at -20°C. |
| Precautions | This product is for research use only. Not for use in diagnostic or therapeutic procedures. |
| Name | MOGS (HGNC:24862) |
|---|---|
| Function | In the context of N-glycan degradation, cleaves the distal alpha 1,2-linked glucose residue from the Glc(3)Man(9)GlcNAc(2) oligosaccharide precursor in a highly specific manner. |
| Cellular Location | Endoplasmic reticulum membrane; Single-pass type II membrane protein {ECO:0000250|UniProtKB:O88941} |

Thousands of laboratories across the world have published research that depended on the performance of antibodies from Abcepta to advance their research. Check out links to articles that cite our products in major peer-reviewed journals, organized by research category.
info@abcepta.com, and receive a free "I Love Antibodies" mug.
Provided below are standard protocols that you may find useful for product applications.
Background
GCS1 cleaves the distal alpha 1,2-linked glucose residue from the Glc(3)Man(9)GlcNAc(2) oligosaccharide precursor in a highly specific manner. Defects in GCS1 are the cause of type IIb congenital disorder of glycosylation (CDGIIb). This syndrome is also known as glucosidase I deficiency and is characterized by marked generalized hypotonia and hypomotility of the neonate, dysmorphic features, including a prominent occiput, short palpebral fissures, retrognathia, high arched palate, generalized edema, and hypoplastic genitalia. Symptoms include hepatomegaly, hypoventilation, feeding problems and seizures. The clinical course is progressive and survival is at most a few months.
References
Volker, C., et al., Glycobiology 12(8):473-483 (2002).De Praeter, C.M., et al., Am. J. Hum. Genet. 66(6):1744-1756 (2000).Kalz-Fuller, B., et al., Eur. J. Biochem. 231(2):344-351 (1995).Kalz-Fueller, B., et al., Eur. J. Biochem. 249, 912-912 (1997).
If you have used an Abcepta product and would like to share how it has performed, please click on the "Submit Review" button and provide the requested information. Our staff will examine and post your review and contact you if needed.
If you have any additional inquiries please email technical services at tech@abcepta.com.
Ordering Information
Other Products
Shipping Information
