


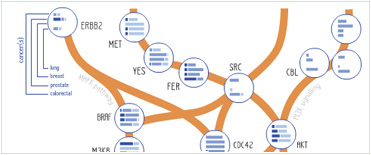







 Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them.
Foundational characteristics of cancer include proliferation, angiogenesis, migration, evasion of apoptosis, and cellular immortality. Find key markers for these cellular processes and antibodies to detect them. The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle.
The SUMOplot™ Analysis Program predicts and scores sumoylation sites in your protein. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle. The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
The Autophagy Receptor Motif Plotter predicts and scores autophagy receptor binding sites in your protein. Identifying proteins connected to this pathway is critical to understanding the role of autophagy in physiological as well as pathological processes such as development, differentiation, neurodegenerative diseases, stress, infection, and cancer.
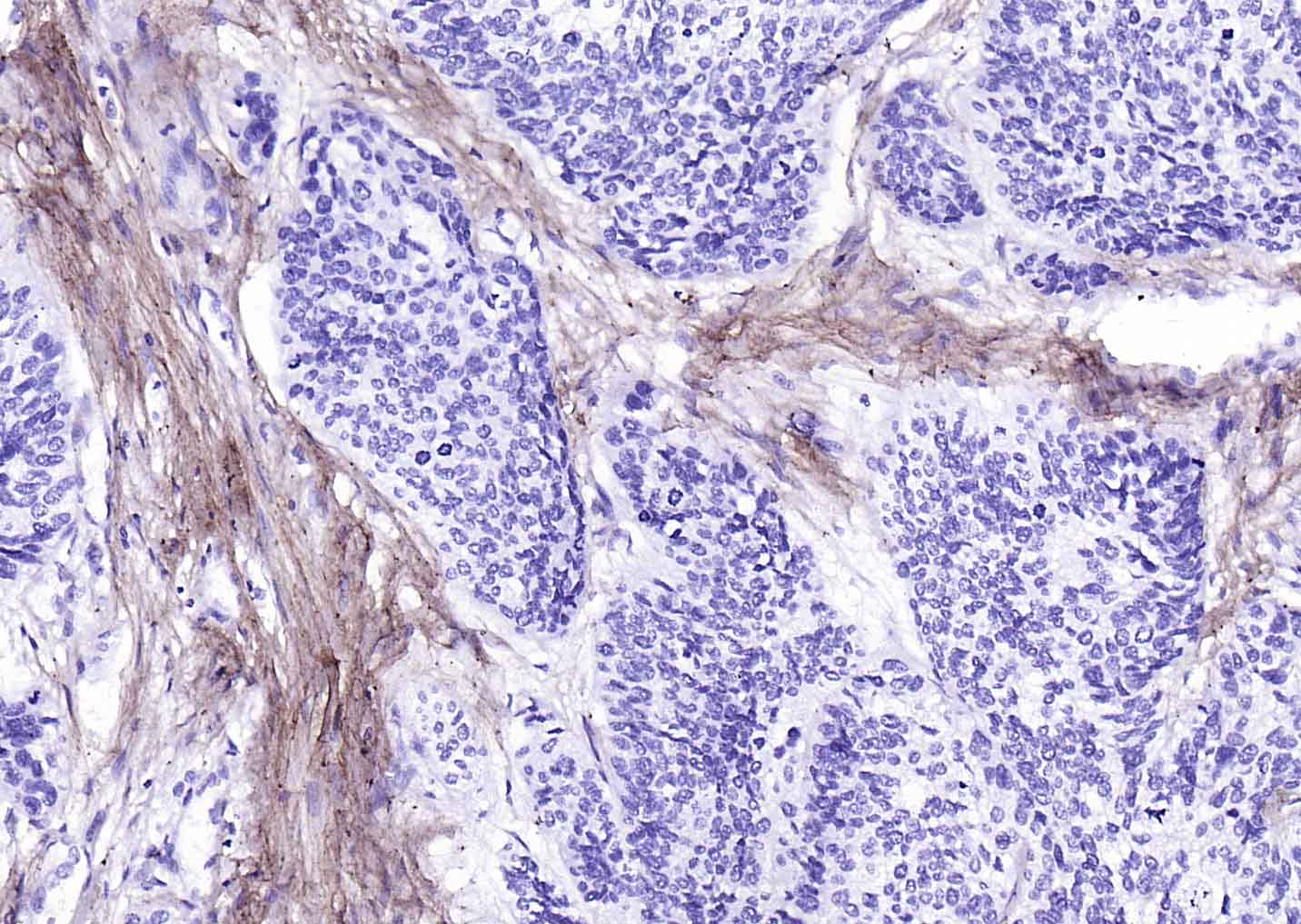
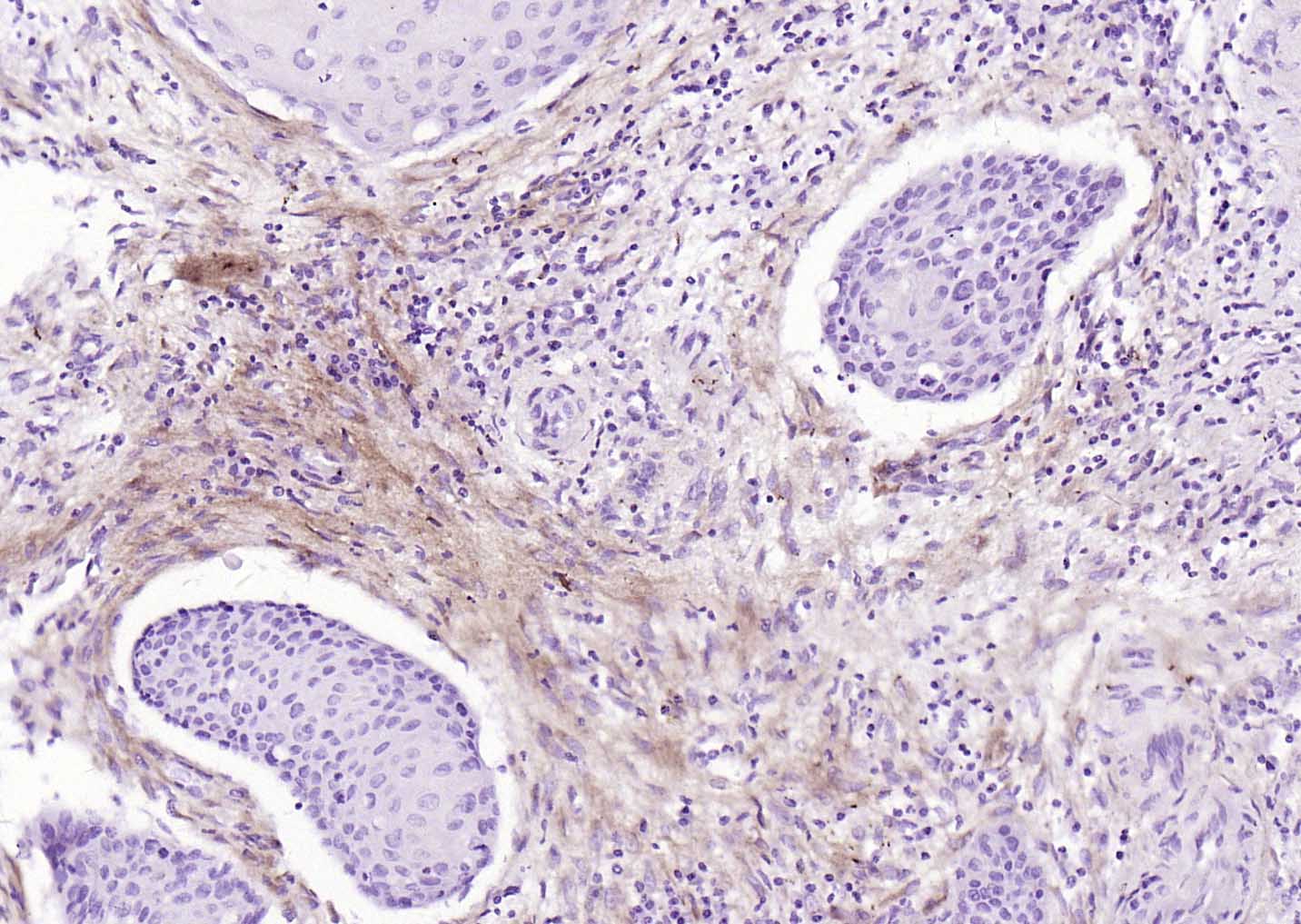
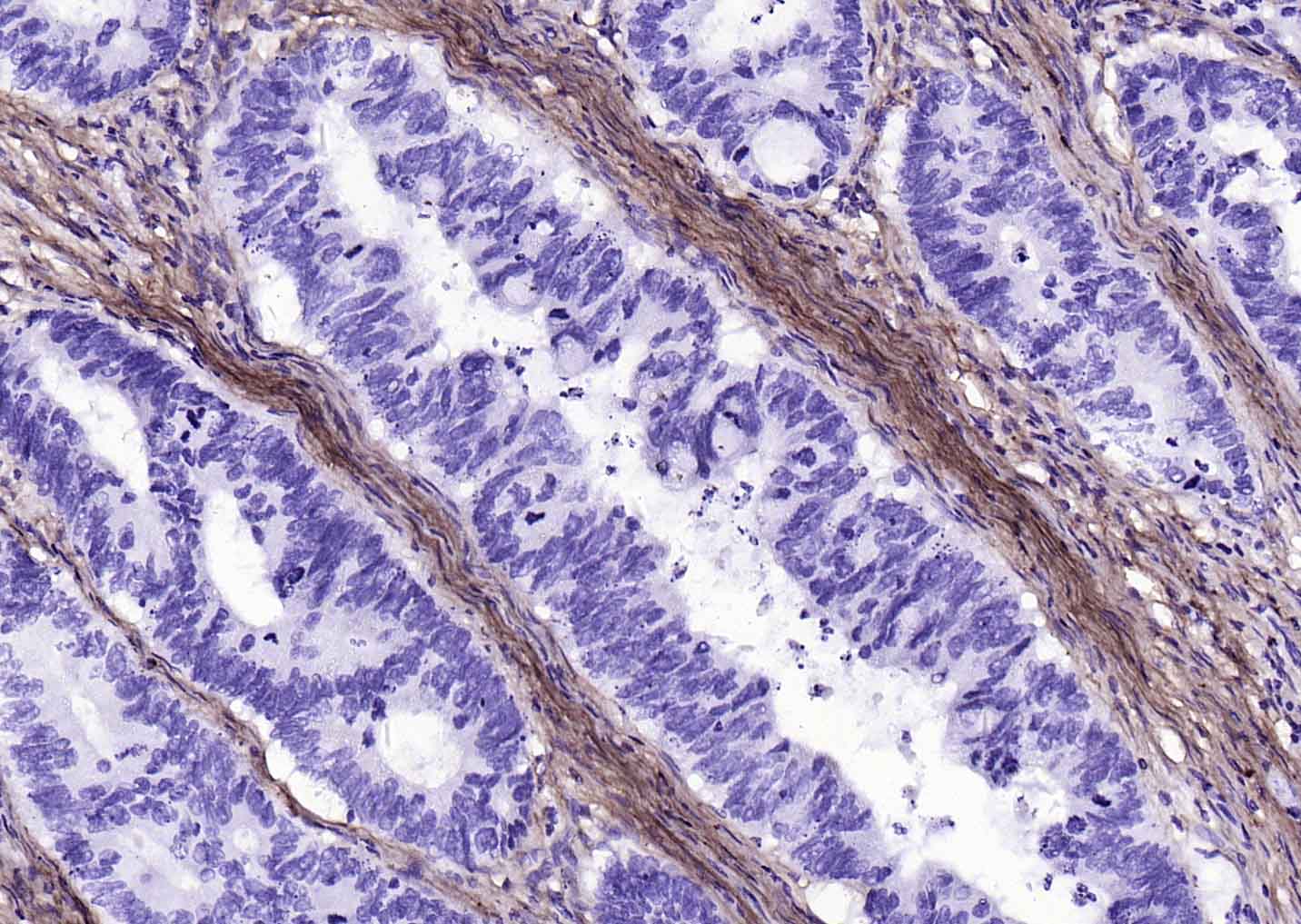
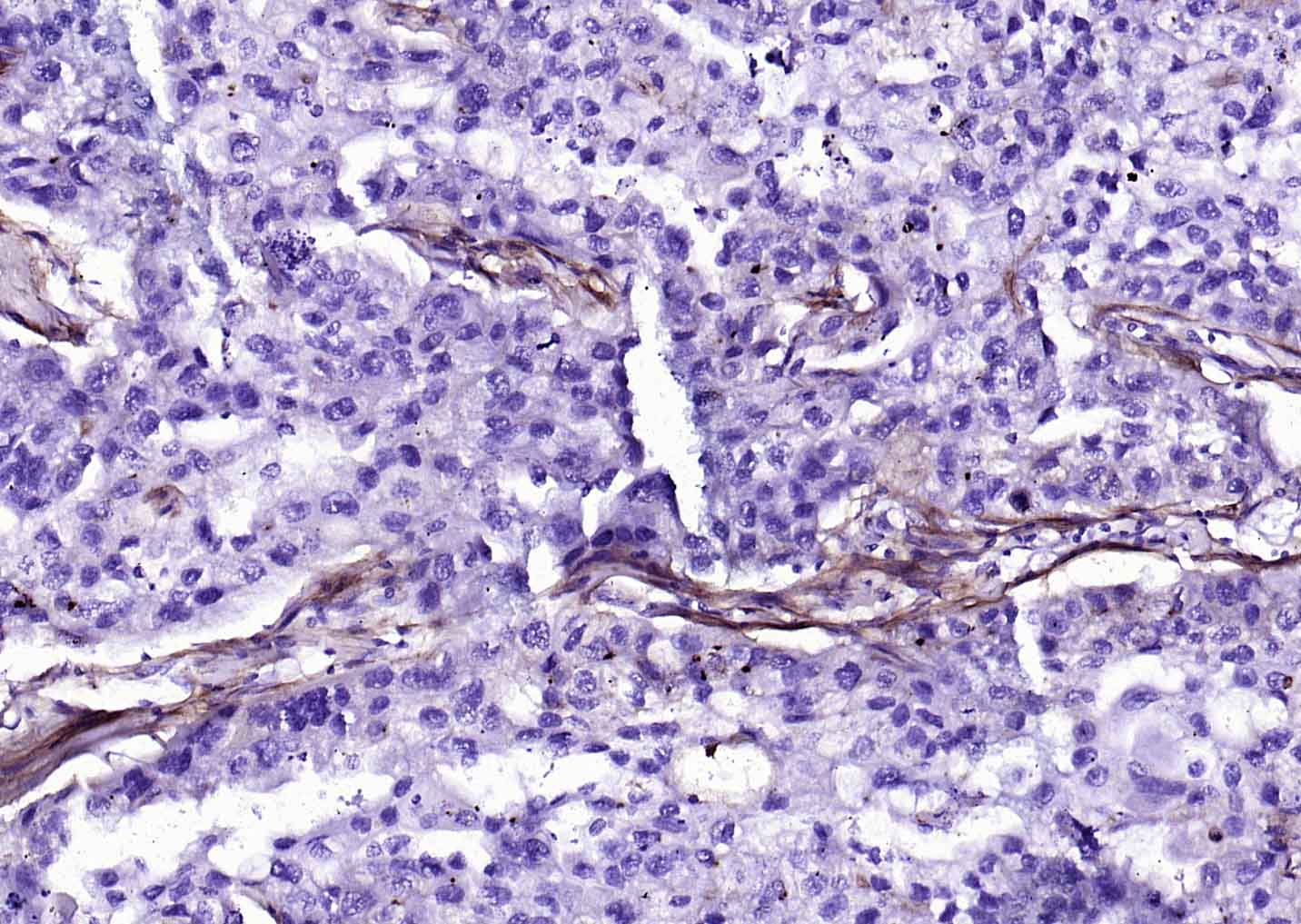
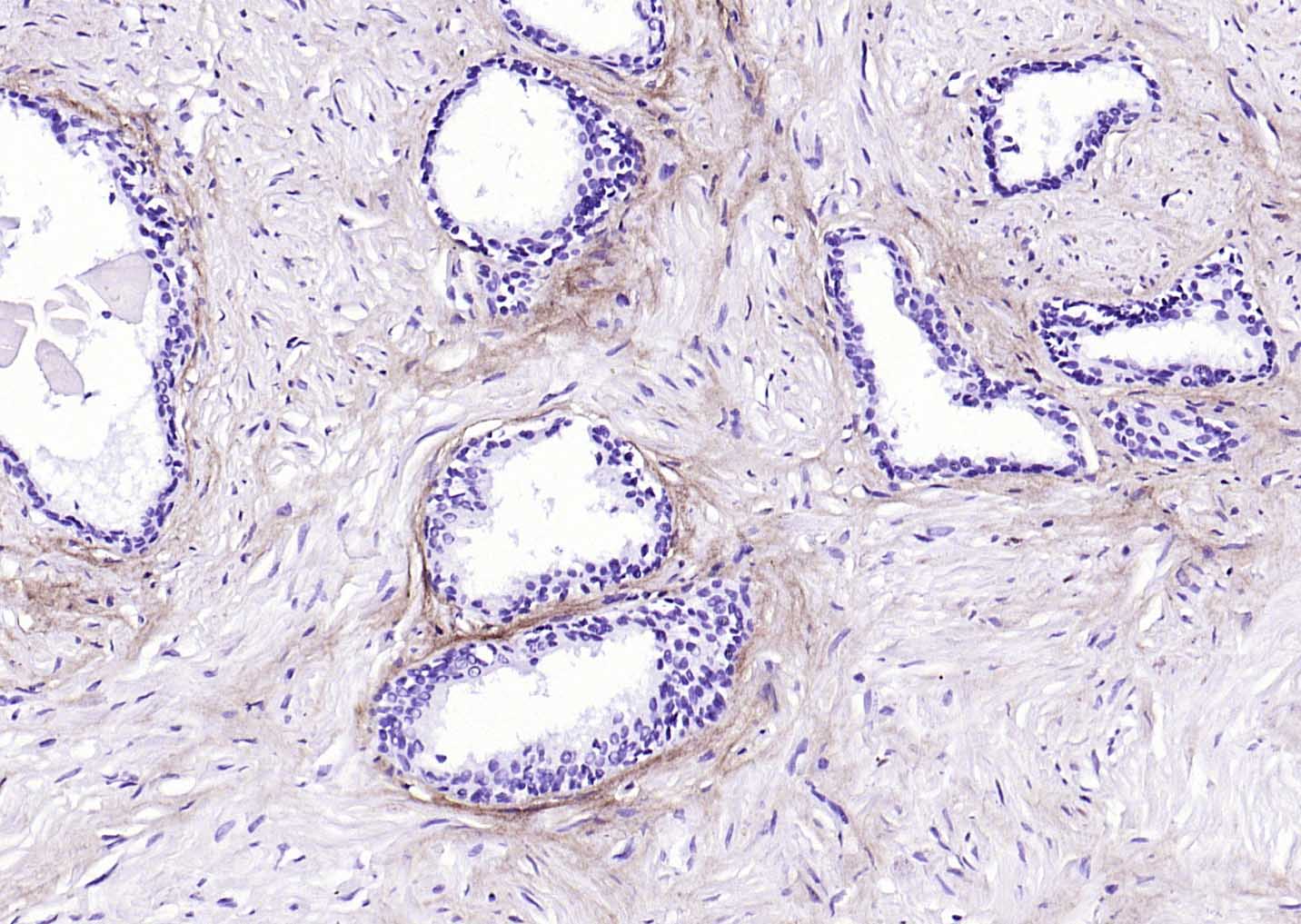
Collagen I Recombinant Rabbit mAb
Collagen I Recombinant Rabbit mAb
- SPECIFICATION
- CITATIONS
- PROTOCOLS
- BACKGROUND

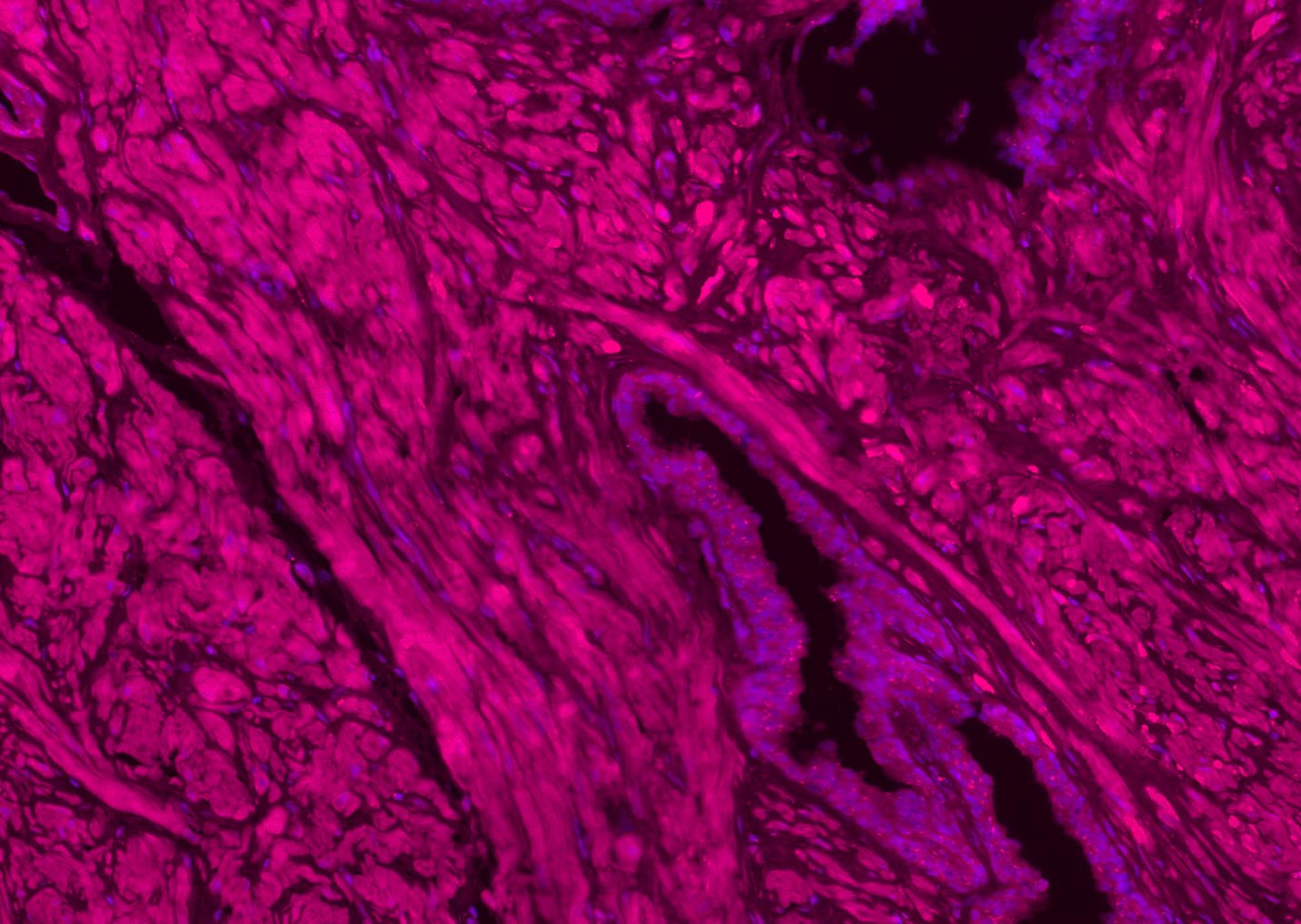
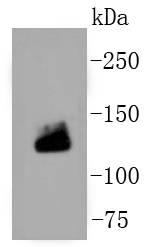
Application
| WB, IHC-P, IHC-F, IF |
|---|---|
| Primary Accession | P02452 |
| Reactivity | Human |
| Host | Rabbit |
| Clonality | Recombinant |
| Calculated MW | 130 KDa |
| Physical State | Liquid |
| Immunogen | KLH conjugated synthetic peptide derived from human Collagen I |
| Isotype | IgG |
| Purity | affinity purified by Protein A |
| Buffer | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| SUBCELLULAR LOCATION | Secreted, extracellular space, extracellular matrix. |
| SIMILARITY | Belongs to the fibrillar collagen family. Contains 1 fibrillar collagen NC1 domain. Contains 1 VWFC domain. |
| SUBUNIT | Trimers of one alpha 2(I) and two alpha 1(I) chains. Interacts with MRC2. Interacts with TRAM2. Subcellular Location : Secreted, extracellular space, extracellular matrix. |
| Post-translational modifications | Proline residues at the third position of the tripeptide repeating unit (G-X-P) are hydroxylated in some or all of the chains. Proline residues at the second position of the tripeptide repeating unit (G-P-X) are hydroxylated in some of the chains. O-linked glycan consists of a Glc-Gal disaccharide bound to the oxygen atom of a post-translationally added hydroxyl group. |
| DISEASE | Defects in COL1A1 are the cause of Caffey disease (CAFFD) [MIM:114000]; also known as infantile cortical hyperostosis. Caffey disease is characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age. Defects in COL1A1 are a cause of Ehlers-Danlos syndrome type 1 (EDS1) [MIM:130000]; also known as Ehlers-Danlos syndrome gravis. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS1 is the severe form of classic Ehlers-Danlos syndrome. Defects in COL1A1 are the cause of Ehlers-Danlos syndrome type 7A (EDS7A) [MIM:130060]; also known as autosomal dominant Ehlers-Danlos syndrome type VII. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS7A is marked by bilateral congenital hip dislocation, hyperlaxity of the joints, and recurrent partial dislocations. Defects in COL1A1 are a cause of osteogenesis imperfecta type 1 (OI1) [MIM:166200]. A dominantly inherited connective tissue disorder characterized by bone fragility and blue sclerae. Osteogenesis imperfecta type 1 is non-deforming with normal height or mild short stature, and no dentinogenesis imperfecta. Defects in COL1A1 are a cause of osteogenesis imperfecta type 2 (OI2) [MIM:166210]; also known as osteogenesis imperfecta congenita. A connective tissue disorder characterized by bone fragility, with many perinatal fractures, severe bowing of long bones, undermineralization, and death in the perinatal period due to respiratory insufficiency. Defects in COL1A1 are a cause of osteogenesis imperfecta type 3 (OI3) [MIM:259420]. A connective tissue disorder characterized by progressively deforming bones, very short stature, a triangular face, severe scoliosis, grayish sclera, and dentinogenesis imperfecta. Defects in COL1A1 are a cause of osteogenesis imperfecta type 4 (OI4) [MIM:166220]; also known as osteogenesis imperfecta with normal sclerae. A connective tissue disorder characterized by moderately short stature, mild to moderate scoliosis, grayish or white sclera and dentinogenesis imperfecta. Genetic variations in COL1A1 are a cause of susceptibility to osteoporosis (OSTEOP) [MIM:166710]; also known as involutional or senile osteoporosis or postmenopausal osteoporosis. Osteoporosis is characterized by reduced bone mass, disruption of bone microarchitecture without alteration in the composition of bone. Osteoporotic bones are more at risk of fracture. Note=A chromosomal aberration involving COL1A1 is found in dermatofibrosarcoma protuberans. Translocation t(17;22)(q22;q13) with PDGF. |
| Important Note | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| Background Descriptions | Collagens are highly conserved throughout evolution and are characterised by an uninterrupted "Glycine X Y" triplet repeat that is a necessary part of the triple helical structure. Type I collagen (95 kDa) is found in bone, cornea, skin and tendon. Mutations in the encoding gene are associated with osteogenesis imperfecta, Ehlers Danlos syndrome, and idiopathic osteoporosis. Reciprocal translocations between chromosomes 17 and 22, where this gene and the gene for Platelet-derived growth factor beta are located, are associated with a particular type of skin tumor called dermatofibrosarcoma protuberans, resulting from unregulated expression of the growth factor. |
| Gene ID | 1277 |
|---|---|
| Other Names | Collagen alpha-1(I) chain, Alpha-1 type I collagen, COL1A1 |
| Target/Specificity | Forms the fibrils of tendon, ligaments and bones. In bones the fibrils are mineralized with calcium hydroxyapatite. |
| Dilution | WB=1:500-2000,IHC-P=1:100-500,IHC-F=1:100-500,IF=1:100-500 |
| Storage | Store at -20 ℃ for one year. Avoid repeated freeze/thaw cycles. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 ℃. |
| Name | COL1A1 |
|---|---|
| Function | Type I collagen is a member of group I collagen (fibrillar forming collagen). |
| Cellular Location | Secreted, extracellular space, extracellular matrix {ECO:0000255|PROSITE-ProRule:PRU00793} |
| Tissue Location | Forms the fibrils of tendon, ligaments and bones. In bones the fibrils are mineralized with calcium hydroxyapatite |

Thousands of laboratories across the world have published research that depended on the performance of antibodies from Abcepta to advance their research. Check out links to articles that cite our products in major peer-reviewed journals, organized by research category.
info@abcepta.com, and receive a free "I Love Antibodies" mug.
Provided below are standard protocols that you may find useful for product applications.
Background
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
If you have used an Abcepta product and would like to share how it has performed, please click on the "Submit Review" button and provide the requested information. Our staff will examine and post your review and contact you if needed.
If you have any additional inquiries please email technical services at tech@abcepta.com.
Ordering Information
Other Products
Shipping Information
